WHAT ARE CILIA?
Discovery of cilia
When Antonie van Leeuwenhoek, a Dutch tradesman, first looked through a microscope of his own invention, he did not foresee the world that would unveil before him. He was simply interested in the quality of his fabrics. van Leeuwenhoek, was the first man to observe microorganisms swimming around, as early as 1674, which he described as ‘little animals with diverse incredibly thin little feet, or little legs, which were moved very nimbly...and wherewith they brought off incredible quick motions’.These little legs are now known as motile cilia, and have since then been a topic of intense investigation. However, it was not until the 1900s that scientists reported the existence of non-motile, solitary, primary cilia and began to explore their function. In the following 100 years the scientific community slowly gained insights in the structure and function of this intriguing cell organelle, but a major leap in the understanding of primary cilia was only made in the last 20 years when advances in molecular genetics linked mutations in genes that encode ciliary proteins with several hereditary syndromes. The phenotypes of those syndromes although diverse proved to all lead back to defects in cilia which resulted in the identification of a new class of inherited diseases now collectively termed ciliopathies.
This class comprises diseases which had already been known such as Bardet-Biedl syndrome (BBS; first described around 1900, later clinically specified), Joubert syndrome (JBTS; first described in 1969) and Nephronophthisis (NPHP; first described in 1951). Patients suffer from obesity combined with blindness (BBS), neurodevelopmental defects (JBTS) and/or kidney failure (NPHP). Because of this broad presentation of clinical features, they were never identified as phenotypes arising from malfunctioning of the same organelle, the primary cilium.
Cilia are cellular organelles with a slender, longitudinal shape that protrude from the cellular membrane of almost all cell types. Two main types of cilia can be distinguished, motile cilia and immotile cilia. Motile cilia often occur as bundles of up to 200-300 hair-like cilia on the cell surface, that are capable of beating in a constant frequency whereby a fluid is propelled as observed in protozoan ciliates by Antonie van Leeuwenhoek). In cells with immotile or primary cilia, however, only one cilium is generated per cell, and this single cilium has evolved as the cell’s communication center, which allows exchange of molecular information with its surroundings.
Cilia Structure
Cilia project from a basal body, a centriole-based structure composed in most vertebrate cells of nine triplets of microtubules, arranged in a nine-fold symmetry, that often become doublets towards the distal ends. These ends are used as a foundation to build the ciliary microtubule skeleton, the axoneme. The axoneme is composed of nine doublets of microtubules, tubular polymers of tubulin, which are tightly wrapped by the ciliary membrane. The plus ends of the microtubules, where growth by polymerization or treadmilling takes place, are situated at the tip of the cilium. The region directly apical to the basal body is specified as the transition zone (TZ), which functions as a gate and regulates the entry and exit of proteins into and out of the cilium. Structurally, the TZ can be recognized by Y-shaped linkers between the axoneme and the ciliary membrane, which together form the ‘ciliary necklace’, a term originating from the similarity of a beaded necklace wrapped around the base of the cilium (Figure 1). Just below the TZ, transition fibers are connecting the basal body to the membrane. These structures are proposed to have a dual function. Firstly, they serve as a physical diffusion barrier for vesicles targeted towards the cilium. Transmembrane proteins, which arrive in vesicles at the TZ, are forced to integrate into the ciliary membrane, after which they will be transported (still being within the plane of the ciliary membrane) towards the tip of the cilium by the intraflagellar transport (IFT) proteins. Vesicle delivery to the cilium contributes to the establishment of a highly specialized ciliary membrane that contains various receptors and signaling proteins. The other function of transition fibers is to provide ample docking and assembly sites for the IFT proteins (Figure 1). IFT is essential for ciliary function as protein synthesis is not believed to occur within the cilium itself. There are two types of IFT complexes which bind specific motor proteins to transport proteins in an anterograde direction, from the base to the tip of the cilium (IFT-B powered by cytoplasmic motor dynein-2), or in a retrograde direction, back from the tip to the base (IFT-A powered by the kinesin-2 motor). Finally, there is an invagination of membrane at the base of the cilium, called the ciliary pocket (Figure 1), which has been proposed as a major site of exocytosis and endocytosis of ciliary proteins.
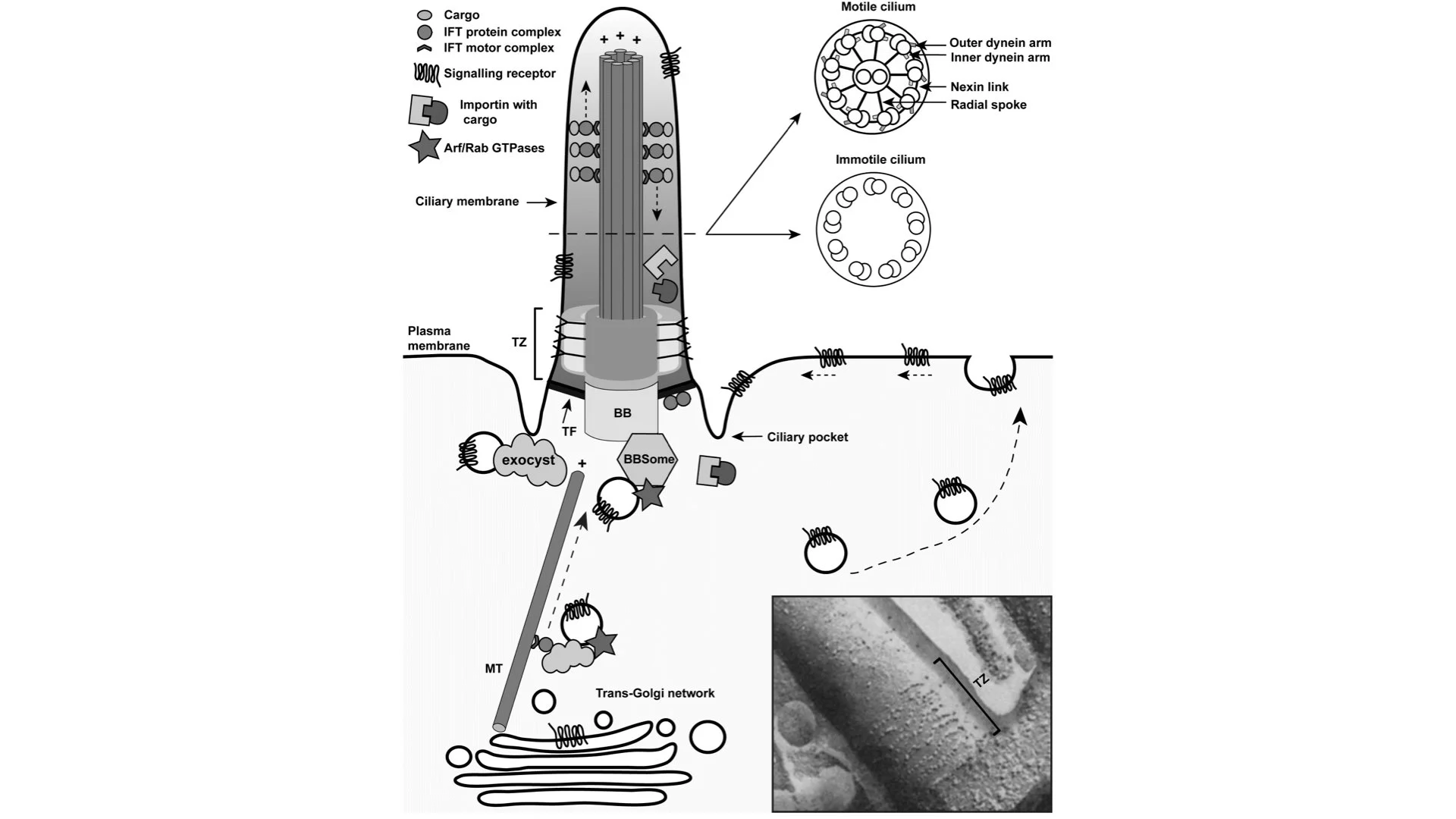
Figure 1.
Schematic structure of the cilium. Cilia are composed of a basal body (BB) and a ciliary axoneme that protrudes from the apical plasma membrane. These two structures are linked via the transition zone (TZ) that consists of three compartments, i.e. a base, and two compartments filling the space between the microtubules and the axonemal membrane. Structurally the TZ is defined by the presence of Y-shaped linkers, which together form the ‘ciliary necklace’ that is wrapped around the base of the cilium, as shown in the electron microscopy image at the bottom right corner. The TZ acts as a diffusion barrier and regulates ciliogenesis and signaling. Cilia contain a microtubule cytoskeleton that is composed of nine microtubule doublets organized in a ring, with the plus-ends situated at the ciliary tip. A central microtubule pair is present only in motile cilia. These microtubules together with inner and outer dynein arms, radial spokes, and nexin links drive ciliary movement. To generate movement dynein heavy chains of one doublet slide against microtubules of a neighboring doublet thereby orchestrating the beating of the cilium in an ATP-dependent fashion. In both cilium types the ciliary microtubules
Cilia Function
The antenna-like appearance of the primary cilium reveals its function: cilia sense environmental cues coming from other cells or from outside the human body. The signals are received by ciliary receptors, converted into an intracellular signal, and subsequently transduced to the cell body. Because of their function in several developmental signaling pathways, cilia play a pivotal role in vertebrate development. Examples of cilia-dependent pathways are platelet-derived growth factor signaling (e.g. involved in chemotactic-directed migration of fibroblasts), Notch signaling (cell proliferation and differentiation), Hippo pathway (regulating cell proliferation, survival, and differentiation) and Wnt signaling (modulating the cytoskeleton). The latter can be divided into two branches, a canonical β-catenin dependent branch and non-canonical branch or planar-cell polarity PCP) pathway. The canonical Wnt pathway has important roles in the formation of neuronal circuits during development and involves stabilization followed by nuclear localization of β- catenin, resulting in transcription of target genes. The cilium is believed to dampen the canonical Wnt pathway by a spatial regulation of Jouberin (AHI1), which normally facilitates the translocation of β-catenin to the nucleus. When Jouberin is retained in the ciliary compartment, it impairs the nuclear localization of β-catenin, thereby modulating the pathway activity. The non-canonical PCP pathway is linked to left-right patterning and neural tube closure and results in changes in cell morphology (actin dynamics, cell polarity, and cell shape) rather than transcription. In more recent years, studies of Wnt-signaling in (defective) cilia have shown contradicting results and therefore the exact relationship with the cilium remains under debate. The best described signaling pathway in cilia is the Sonic Hedgehog (Shh) signaling cascade Figure 2). Defects in the Shh signaling pathway can cause various birth defects, including incomplete separation of the brain hemispheres (holoprosencephaly), polydactyly and craniofacial and skeletal malformations. In studies of mutant animal models with these phenotypes, variants in Shh genes, but also genes encoding IFT machinery have been identified. Furthermore, the link with the cilium became clear when several key players for Shh were localized to the cilium. In the case of correct neural tube development, a ventro- dorsal gradient of Shh protein is essential for a correct spatial differentiation of specific subtypes of neurons. The different concentrations of Shh result in different levels of Shh pathway activity, and thereby control (either activating or repressing, depending on the level) the expression pattern of sets of target genes. This mechanism causes specific patterning of progenitor cells during development, which is essential for the correct formation of the central nervous system, as well as the limbs. Because of the differential expression of ciliary signaling proteins in tissues and cellular subtypes, variants can cause specific disturbances of signaling pathways in a subset of cells and thereby lead to a spectrum of phenotypes.
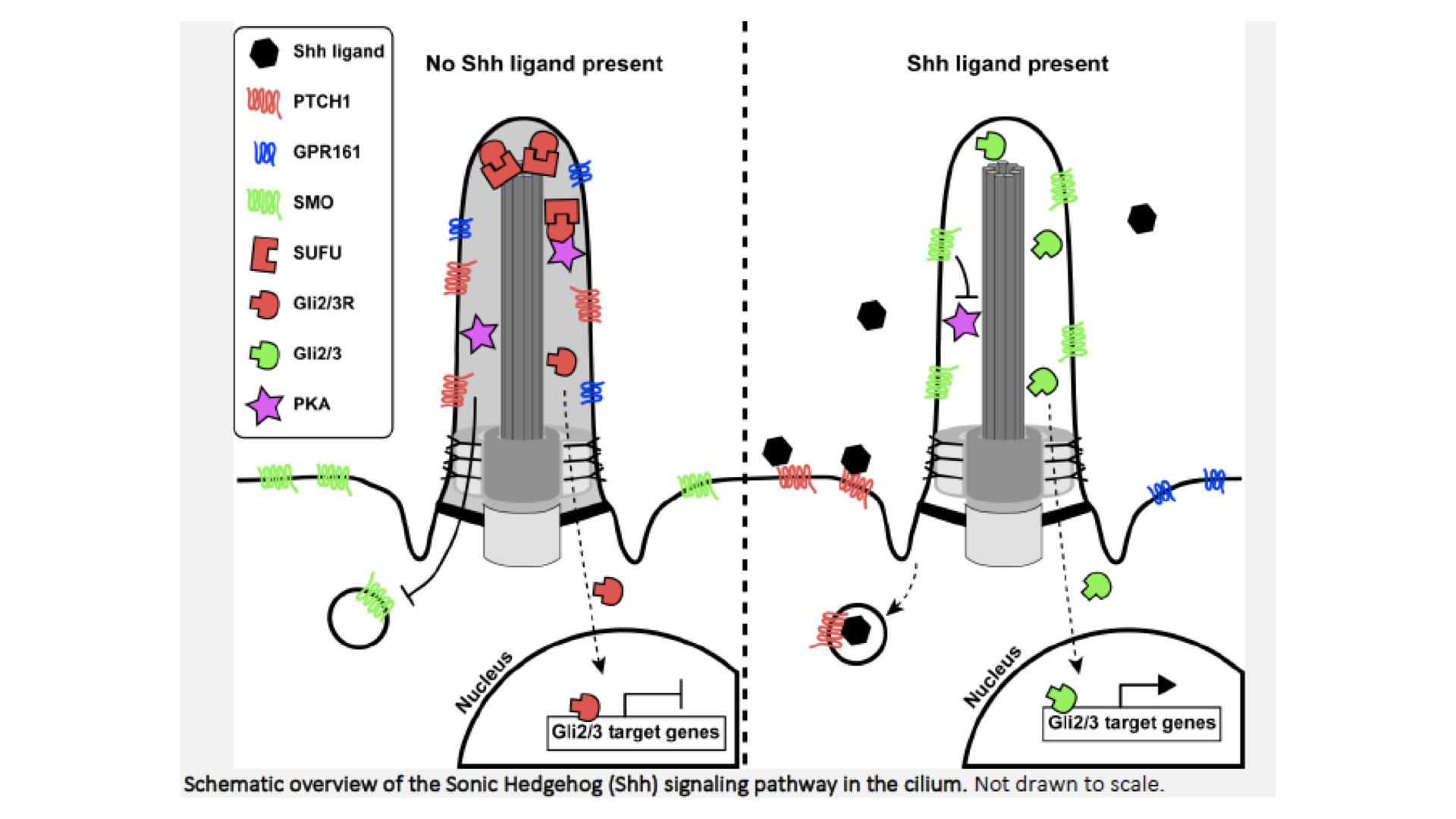
Figure 2.
Shh pathway overview. When no Shh ligand is present, the Patched-1 (PTCH1) receptor represses Smoothened (SMO) from activating Gli2 and Gli3 transcription factors and entry of SMO into the ciliary membrane. Furthermore, the Gli transcription factors are bound by SUFU. When Shh ligand is present, PTCH1 is activated and will release SMO, allowing its ciliary entry and translocation to the tip. This will activate and release the Gli transcription factors from SUFU, allowing them to translocate from the cilium to the nucleus and activate downstream target genes important for neuronal and skeletal development. These Gli transcription factors are bi-functional and can also actively repress transcription. When the pathway is not activated, Gli2 and Gli3 are phosphorylated by PKA, CKI and GSK3β, causing proteolytic cleavage which generates repressor forms (Gli2R and Gli3R). PKA is thought to be activated by increased cAMP levels generated by GPR161. Activation of the Shh pathway promotes the exit of GPR161 from the cilium, and thereby stops Gli2/3 phosphorylation and generates high concentrations of activating (uncleaved) transcription factors. The precise balance of activator and repressor forms of the Gli transcription factors determines the gene expression pattern of the cell and thereby its differentiation into a specific progenitor subtype.